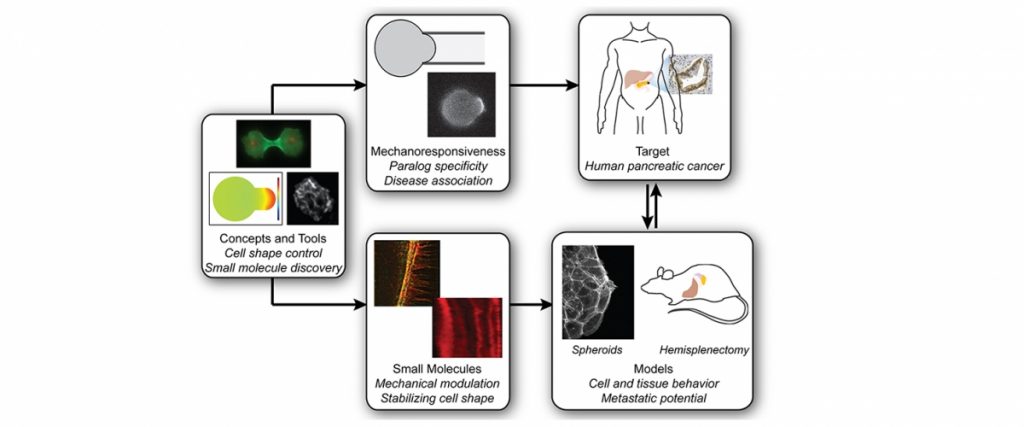
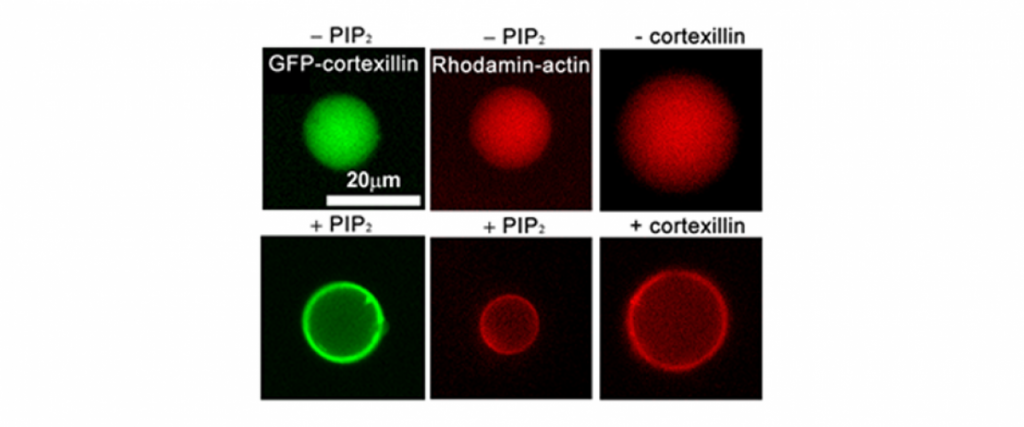
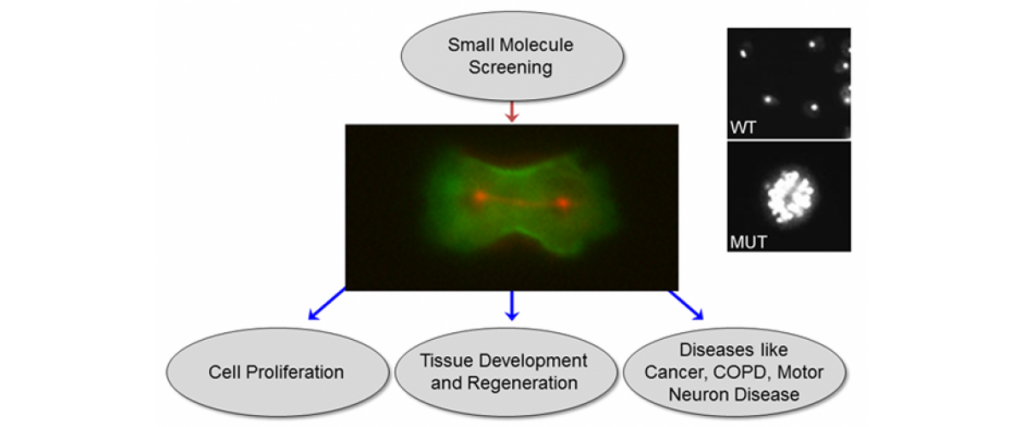
We have created a platform-based approach to small molecule discovery based on the concept that major health challenges, namely cancer metastasis and chronic obstructive pulmonary disease, are associated with defects in control of cell and tissue morphology and mechanics. We bring a unique suite of tools, drug screening strategies, and expertise for identifying and developing novel chemical modulators (future drugs) of cell morphology to attack these major health problems. Most strategies for drug discovery draw upon small molecule screening against specific target proteins. However, these compounds often lead to effects at the cell and organism level that cannot be anticipated. Instead, we recognize that diseases are ultimately manifested in alterations in cell and tissue behaviors. These behaviors are controlled by networks of biomolecules, not any single one. Therefore, we start our drug discovery approaches by identifying small molecules or combinations of molecules that directly modulate these cell and tissue behaviors and then use a variety of genetic, biochemical and biophysical approaches to identify the molecular bases for the compounds’ action. The combination of strategies constitute Biology-In-Action (BIA), which is used to identify our compounds, and then Quantitative Phenotypic Fingerprinting (QPF), which is used to identify the targeted pathways and targets of the compounds. Here we provide a summary of our work in just one of these areas, which is in oncology.
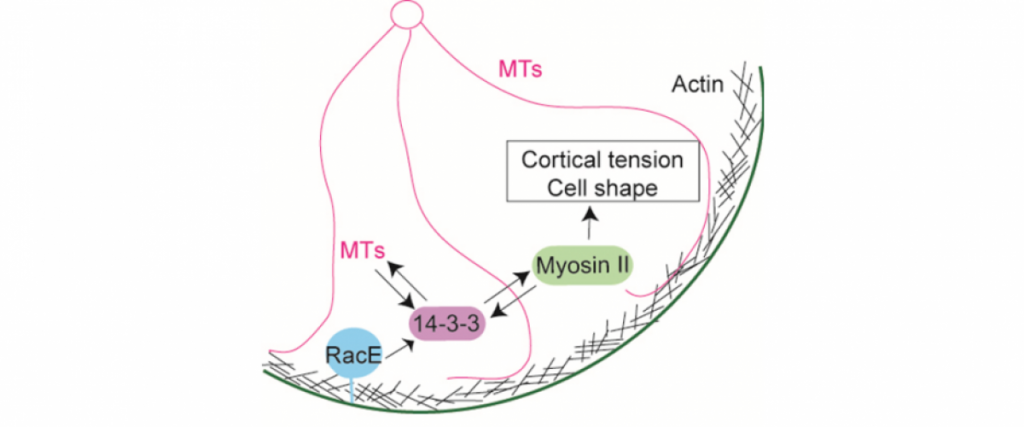
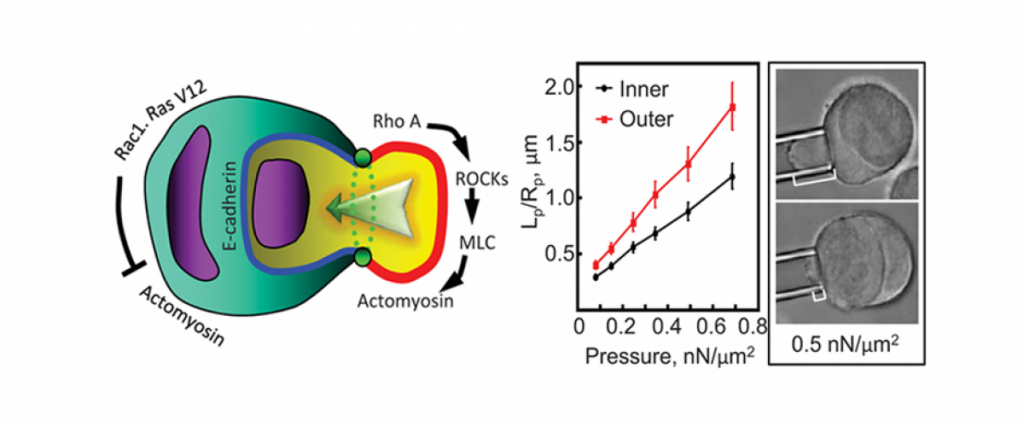
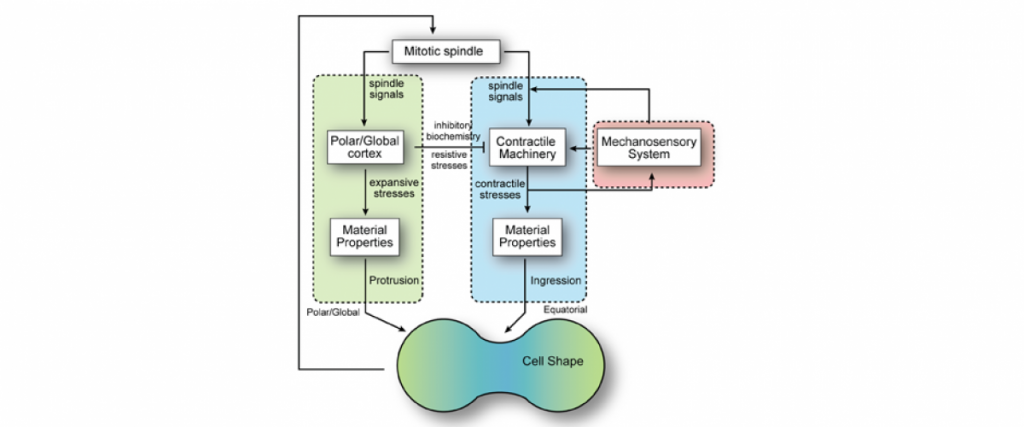
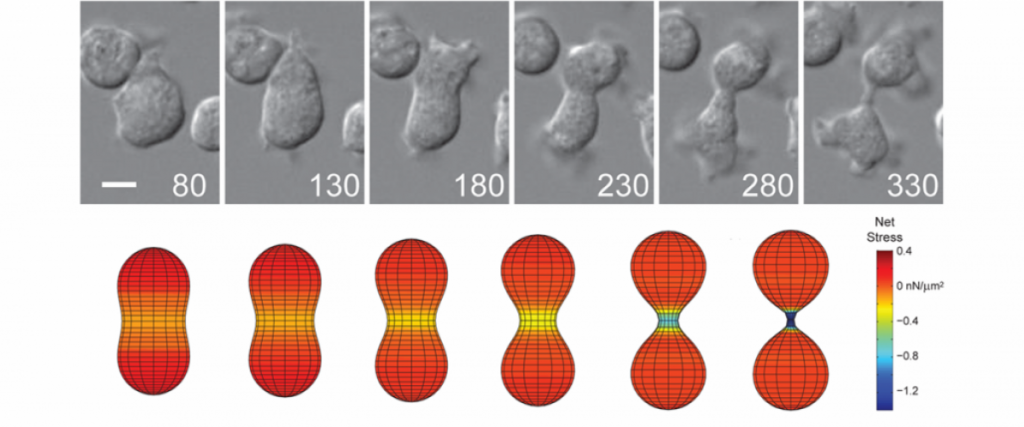
Importantly, for pancreatic cancer (an immediate focus of our work), the major drivers include activated mutant versions of an important signaling protein KRAS, inactive mutant forms of a quality control surveillance protein TP53, as well as a collection of other mutant proteins and proteins with significantly upregulated expression. However, we found that many key proteins (e.g. specific versions of the motor protein myosin II, myosin IIA and IIC) in the cell’s powertrain are significantly upregulated. Even more provocative, myosin IIA can slow and block tumor progression (tumor suppressive function). This motor protein does this by modulating the power output and mechanical state (how stiff or tense the cell is). Through these activities, myosin II activity also feeds back onto quality control proteins like TP53 (much like the speed of the car is fed back to the cruise control system so that engine power is modified to maintain course). However, in cancer, many of the signaling pathways become altered leading to disrupted myosin II motor function. When these systems become disturbed, the cell system is poised to grow uncontrollably, change shape, invade surrounding tissue, and even break free so that the cell or small clusters of cells can move to distant locations and set up new colonies (i.e. metastasize).
With our discovery platform, we have identified a strategy to activate the function of one myosin II form (myosin IIC), which is specifically overexpressed in pancreatic cancer. Using a small molecule (4-HAP), we can increase the amount of functional myosin IIC and correct many of the mechanical defects associated with pancreatic cancer progression. The consequence for the cancer is that the cells have reduced ability to migrate through obstacles, and in animal studies, 4-HAP reduces the ability of the cells to form metastases (Fig. 1). We also hypothesize that in addition to the reduction in the ability of the cancer cells to invade and metastasize, we are pushing the cells back towards a less cancerous state. This collective effect of targeting the powertrain to block metastasis and return cancer cell identity to a more normal state is a new innovative approach to treating cancer.
For more information on this work, please see Surcel et al. PNAS 2015, Surcel and Robinson PNAS 2019, Surcel et al. Cancer Research 2019 , Bryan et al. PNAS 2020 and Parajón et al. AJP Cell Physiol. 2020.